
作者 | 康奈尔大学 Fengqi You 团队
编辑 | ScienceAI
康奈尔大学Fengqi You教授团队提出了结合量子计算与生成式AI的逆向分子设计框架,充分利用了两者的优势。
该框架利用QC辅助的深度学习模型来学习化学空间并模拟其特性,以预测和生成具有特定化学性质的分子结构。生成式AI在整个过程中扮演关键角色,通过学习大量分子数据的潜在结构-性质关系,生成符合预设性质且可合成的新分子候选物。
引入量子计算为化学系统的研究提供了高效的计算能力和优化算法,解决了传统计算机在处理大规模化学系统时的性能瓶颈。通过量子-经典混合计算框架,研究人员能够在复杂的化学空间中进行高效、有效的分子设计,为新分子的发现和材料科学的进步开辟了新途径。这一技术的应用将大大加速化学研究和材料科学的发展,为解决现实世界中的各种问题提供强有力的工具。
相关研究以《Molecular design with automated quantum computing-based deep learning and optimization》为题,发表在《npj Computational Materials》上。
研究背景
分子设计 Vs 逆向分子设计
分子设计是一种创造新分子的过程,它基于现有知识和原理。科学家们根据对化学结构和性质的理解,设计和合成新分子。这个过程涉及从理论上预测新分子的性质,并通过实验来验证。分子设计在药物开发和新材料创造中扮演着重要角色,它利用已知的化学信息来推动新分子的发现。
逆向分子设计(Inverse Molecular Design)则是从所需的性质出发,通过计算方法和人工智能技术在化学空间中搜索或构建能表现这些性质的分子结构。这种数据驱动的方法使得药物发现和功能材料开发更为高效,尤其在需要探索未知化学空间以发现具有特定功能的新分子时更显其优势。逆向设计强调从目标性质出发,逆向推导出可能的分子结构。
相较于传统分子设计,逆向分子设计的优势在于其高度的数据驱动性和计算密集性,允许科学家们在庞大的化学空间中有效地筛选出理想的分子,这在传统方法中是不可想象的。它通过人工智能算法加速了药物筛选和新材料的发现过程,特别是在对未知领域进行探索时,能够发现传统方法可能忽视的结构。这种方法在药物发现和功能材料开发中尤为有用,能够显著提高发现的速度和创新性。
生成式人工智能
随着机器学习以及深度学习技术的发展,生成式人工智能(Generative AI)在逆向分子设计中的应用,展现了其强大的数据处理和学习能力。通过深度学习模型,如生成对抗网络(GANs)和变分自编码器(VAEs),这些AI系统能够理解和模拟分子数据集的复杂分布,生成符合特定性质要求的新分子。
这些模型分别依靠对抗过程和概率编码来生成数据,其中GANs由竞争的生成器和鉴别器组成,VAEs则通过优化潜在空间的表示来重构数据。这一技术大幅提升了在药物设计和新材料发现领域的效率。
尽管如此,生成式AI在实现稳定性和合成可行性方面还存在挑战,并且当数据集质量和量级不足以支持复杂分子结构的学习时,其性能可能受限。因此,尽管生成式AI为逆向分子设计提供了前所未有的工具,但仍需要在算法优化、数据质量提升以及跨学科合作方面进行持续的研究和发展。
研究的主要贡献
1. 提出了一种数据高效的量子-经典混合方法,用于分子性质估计,该方法利用了QC辅助学习训练的深度学习模型来提取分子的稳健潜在表示。
2. 开发了一种基于QC的近似优化技术,利用训练好的性质估计模型以指导的方式探索化学空间并识别具有所需性质的候选分子。
3. 与现有基于深度学习的分子设计方法相比,所提出的基于量子计算辅助的分子设计框架高效地生成了多个具有不同生理化学性质目标的类药物分子。
模型结构
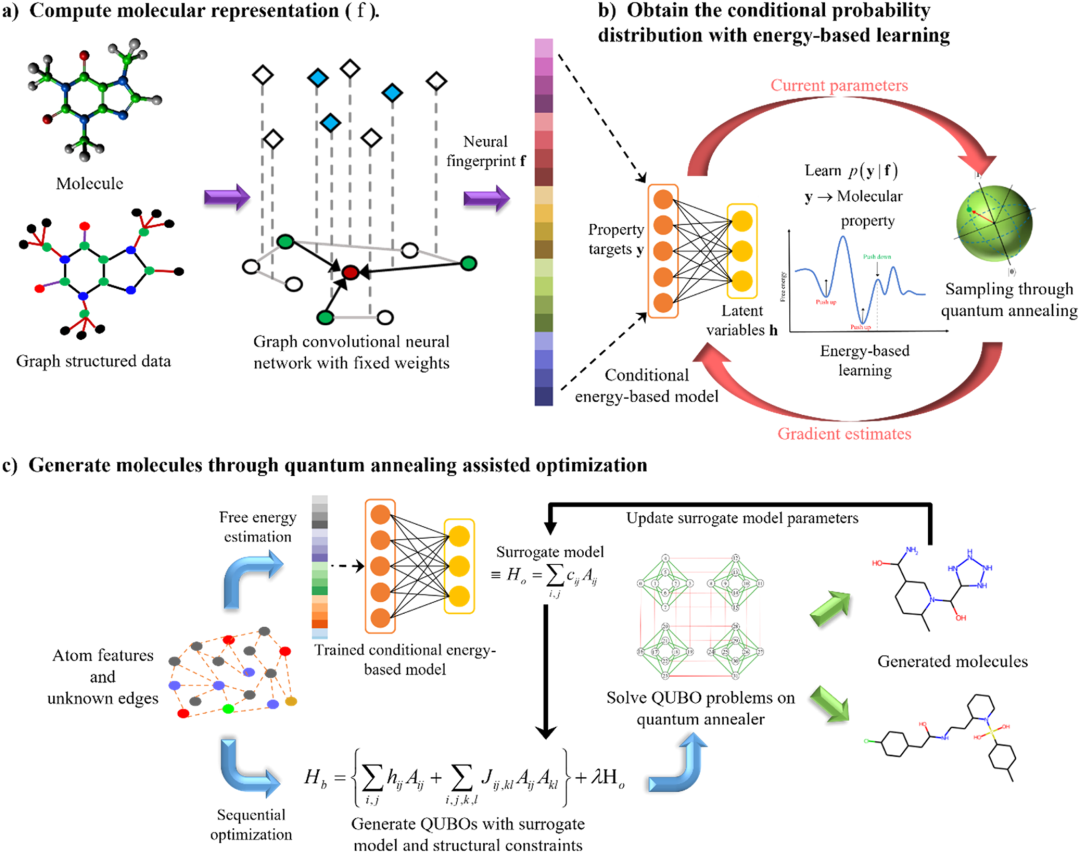
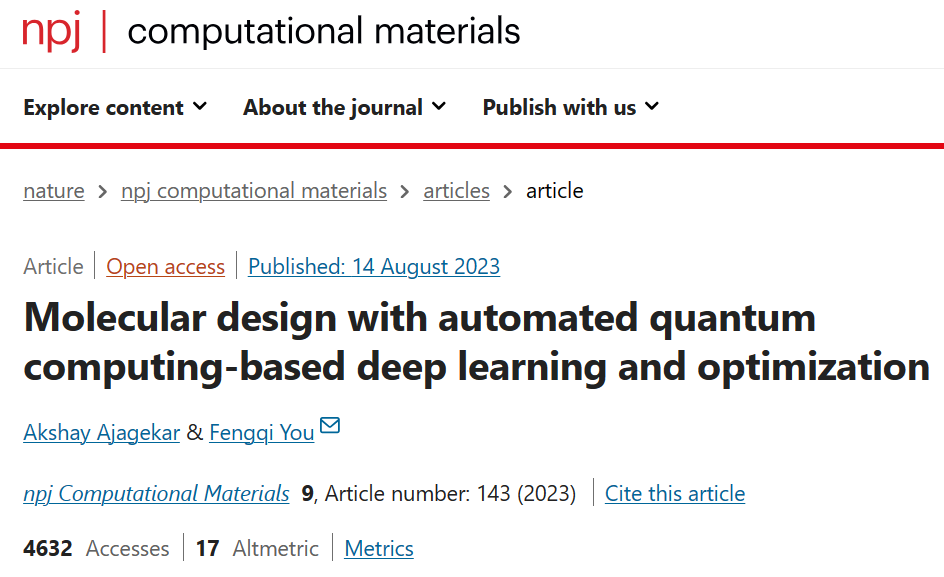
图1概述了本研究中用于分子生成的QC辅助的学习和优化策略。该图展示了基于能量的模型,该模型通过从量子退火器(quantum annealer)抽取样本进行训练,并捕捉分子结构与分子性质之间的关系,然后模型根据习得的关系,通过图卷积神经网络生成具有目标性质的新的分子结构。
此外,另外一个条件能量基模型被用于估计输入分子的自由能并计算目标值。通过量子退火器以迭代方式解决二次无约束二进制优化(QUBO)问题,产生具有所需目标性质的候选分子。
具体实验结果细节如下:
图2中展示了采用不同方法获得的各种输入数据在前馈模型预测性能方面的误差比较结果。
本文通过训练多个条件能量模型,并结合对比发散(CD)学习与QC辅助学习策略,获取了一系列这些潜在表征。利用相应能量模型的每个潜在表征,通过前馈网络进行重复实验,以测量相关指标及其统计特性。
在采用基于规则的分子描述符作为输入的基准预测模型中,较大的ECFP指纹普遍更适合于预测QED和LogP,而含有MACCS的预测模型在预测可及性分数方面表现更佳。
基线模型采用图卷积神经网络模型生成的指纹,在预测分子的药物样性时显示出显著更高的误差,但在其它分子性质方面的预测结果相较于合成可及性评分的最低误差仅增加了13.6%。与此相对,使用经典和QC辅助学习技术训练得到的条件能量模型的潜在变量表示,虽然其维度较低,却在所有属性目标的预测性能上表现出色。
采用QC辅助生成训练得到的潜在表示的预测模型,不仅在预测误差上与其他基线模型相当,而且在预测分子的药物样性时表现出最小的误差。这些计算结果充分证明了通过QC辅助学习训练的条件能量模型所得潜在表示在分子性质预测方面的有效性。
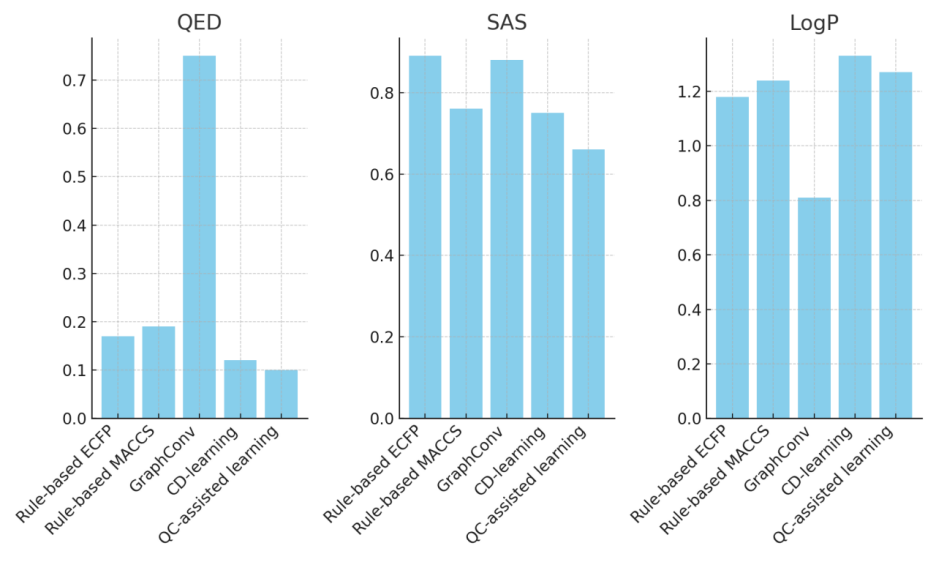
研究者们运用了经过训练的能量基模型,并结合新提出的量子计算(QC)辅助优化技术,对分子的药物样性和分配系数等性质施加了目标条件,实现了有目的的分子生成。
图3展示了这些技术生成分子的统计数据及其计算性质,并与训练集中满足同样条件的分子进行了比较。这一对比揭示了针对特定属性目标,基于QC方法生成的分子确实满足了既定的条件。
然而,某些目标属性下,深度学习方法如CVAE和MGM显示出生成符合要求的分子的能力,而遗传算法GBGA则在这一任务上效率较低,可能需要对每个目标属性进行适应性函数的手动调节。
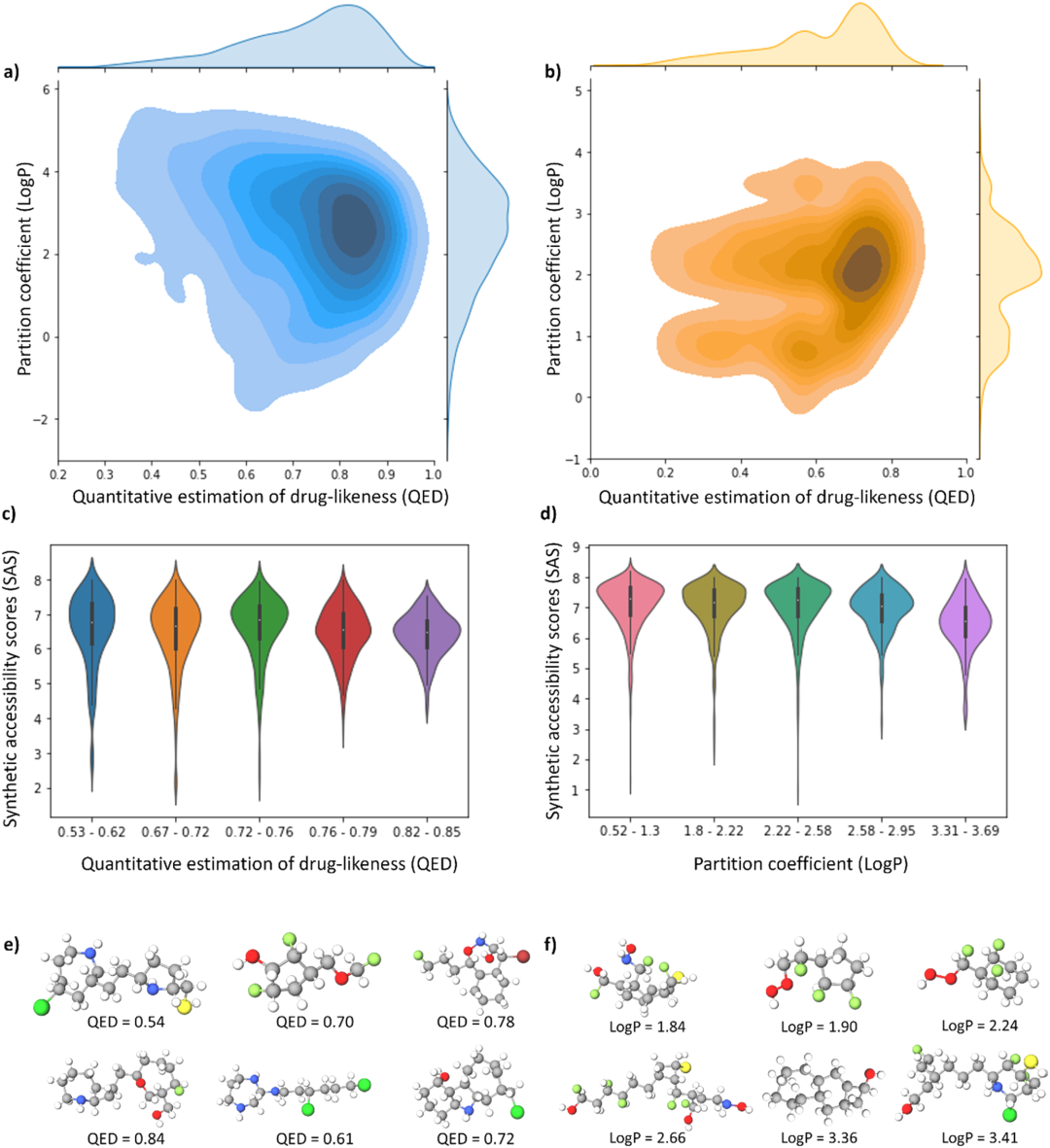
此外,图4中的c和d小图详细描绘了所有生成分子的合成可及性分数(SAS)分布,反映了它们在合成上的难易程度。这些小提琴图表明,QED和LogP值较低的分子在SAS分数上的变异性更大,尽管它们的平均SAS分数相对较高。相反地,药物样性更好和LogP值较高的分子显示出了不同的分布趋势。
研究者还利用t-SNE技术分析了条件能量模型生成的分子的潜在表示,这些通过2D嵌入可视化的表示帮助识别分子与其性质之间的关联度。
图5展现了基于QED值进行着色的训练集分子和生成分子的2D t-SNE嵌入,以及不同属性目标的分子结构示例,显示出不同属性范围内潜在表示的区分,这表明所构建的模型能有效地捕捉分子属性之间的复杂关系。
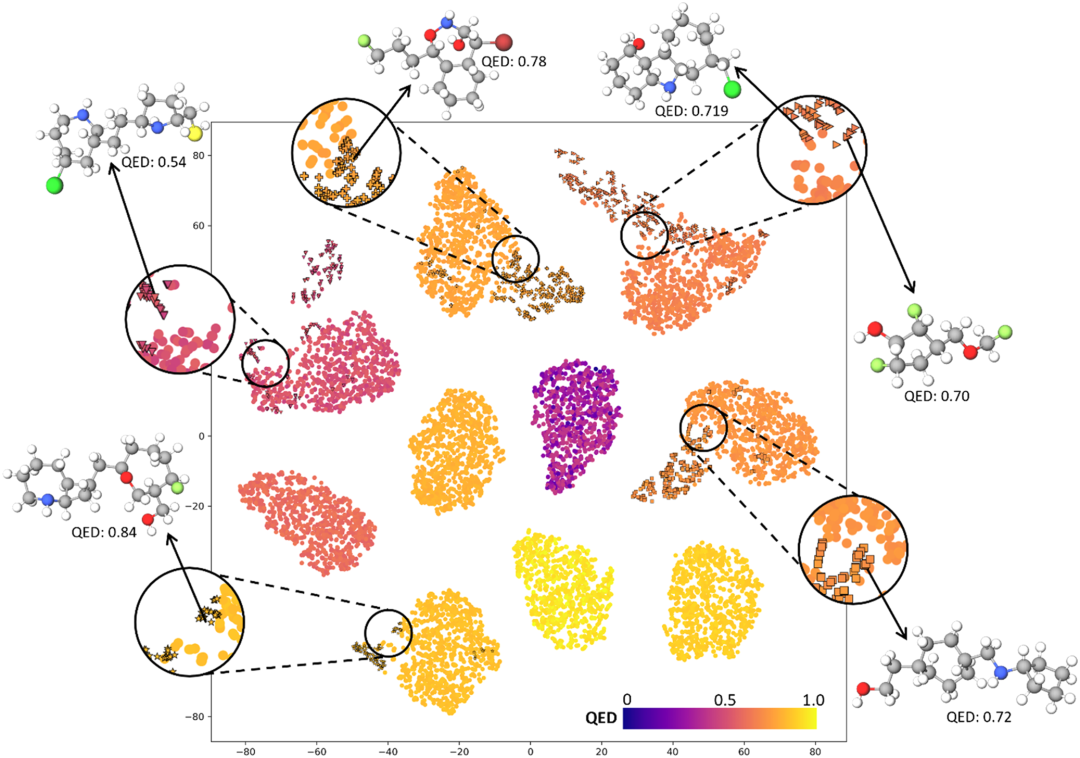
本研究还采用t-SNE嵌入方法来分析使用训练过的条件能量模型生成的分子的潜在表示。通过将这些潜在表示映射到二维空间,有助于揭示分子与其相应性质之间的关联程度。
图5展示了使用t-SNE方法得到的训练集中分子的二维嵌入,以及根据QED属性值进行着色的生成分子。作者在此图中还包括了不同属性目标的分子结构示例。不同属性范围的潜在表示之间的区分表明,具有相似QED值的分子在空间上相互邻近,这表明构建的基于能量的模型成功捕捉了分子与其属性之间的关系,因为具有相似性质的分子在嵌入空间中聚集。



